当化学家们还在为DFT计算中”精度与成本不可兼得”的困境挣扎时,微软用276K参数的神经网络撬动了量子化学的根基
在药物分子筛选的深夜,计算化学家小王盯着屏幕上跳动的数字叹气——传统DFT计算要么精度不够,要么耗时太长。这个困扰领域60年的难题,如今被微软研究院的Skala项目撕开了一道裂隙:他们用深度学习重新定义了交换关联(XC)泛函,让半局域成本的计算精度直逼昂贵的混合泛函。

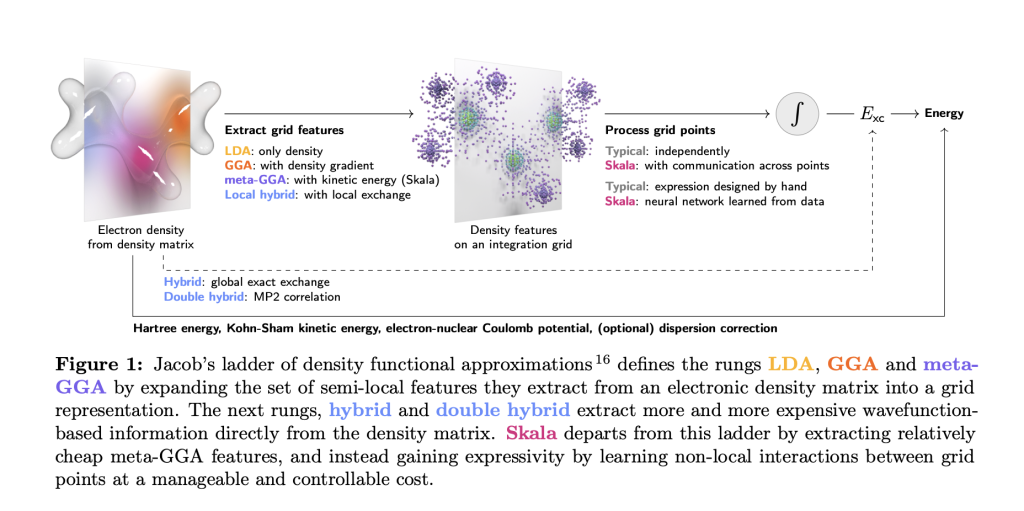
雅各布梯上的革命者
传统DFT计算如同攀登”雅各布阶梯”:从LDA到GGA再到meta-GGA,每提升一阶精度都要付出指数级增长的算力成本。而Skala的突破在于用神经网络学习非局部量子效应,巧妙绕过了传统阶梯的物理限制。
# 感受半局域成本下的混合泛函精度
from pyscf import gto
from skala.pyscf import SkalaKS
mol = gto.M(atom="H 0 0 0; H 0 0 1.4", basis="def2-tzvp")
ks = SkalaKS(mol, xc="skala") # 关键:调用神经网络XC泛函
energy = ks.kernel()
深度学习如何改写物理规则
1. 粗网格点传递的”量子电报”
Skala的创新架构在DFT积分网格中引入粗网格点作为信息中继站(图2)。当处理含380原子的体系时,这些原子中心的”通讯员”让网格点间的信息传递效率提升10倍,却只增加0.1%的计算量。
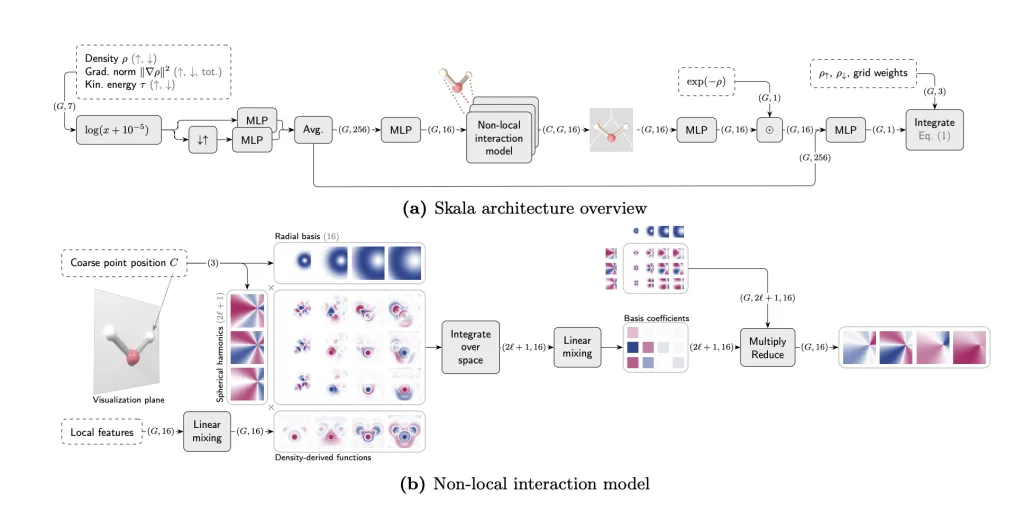

2. 两阶段训练破解密度难题
-
预训练阶段:用B3LYP密度作为”翻译官”,将8万+CCSD(T)/CBS级原子化能数据转化为神经网络可理解的信号 -
SCF微调:让模型在自洽场循环中自我修正,避免陷入”能量-密度”的误差补偿陷阱
正如论文作者在arXiv中强调:”我们首次证明学习非局部性在数据充足时能达到化学精度,且无需手工设计特征”
实测:精度与成本的黄金分割点
在GMTKN55基准测试中,Skala以WTMAD-2 3.89 kcal/mol的成绩逼近顶级混合泛函ωB97M-V(3.23),而计算成本却与meta-GGA相当:
| 测试集 | Skala误差 | ωB97M-V误差 | 计算成本倍数 |
|---|---|---|---|
| W4-17(全) | 1.06 | 2.04 | 0.1x |
| W4-17(单参考) | 0.85 | 1.66 | 0.1x |
| GMTKN55 | 3.89 | 3.23 | 0.1x |
 |

数据来源:Azure NC24ads A100实测,含D3(BJ)色散校正
云原生化学计算新时代
快速部署指南
# 三步启动量子化学加速引擎
pip install torch --index-url https://download.pytorch.org/whl/cu118
pip install microsoft-skala
python -c "from skala.pyscf import SkalaKS; print('GPU就绪:', torch.cuda.is_available())"
Azure AI Foundry上的”一键化学”
微软将Skala集成到云端量子化学平台,支持:
-
批量分子几何优化(含过渡态搜索) -
反应路径能量扫描 -
偶极矩预测(RMSE 5.94 vs B3LYP 7.09)
正如GitHub文档所示:”只需修改
xc="skala"参数,现有PySCF代码即刻获得加速”
常见问题解答
Q:与DM21等ML泛函有何本质区别?
A:Skala放弃手工特征,直接从密度网格学习非局部效应。更重要的是其训练集完全去除了测试集分子,避免数据泄露——这是DM21被诟病的关键缺陷。
Q:何时支持过渡金属?
A:当前版本专注主族元素(H-Ar)。团队在论文中透露,正在用多参考态数据扩展训练集,预计2026年覆盖d轨道体系。
Q:几何优化精度如何?
A:在CCse21基准测试中,键长误差0.012Å(优于B97M-V的0.023Å),但重元素键长预测仍需改进(见HMGB11数据集)。
计算化学的新范式
当Skala在Azure云上以每分子0.3秒的速度完成SCF计算时,我们正在见证计算化学的范式转移:从”物理公式驱动”转向”数据+物理双驱动”。正如微软团队在结论中展望的:”这创造了跨尺度的精度传递链——用波函数精度训练DFT,再用DFT数据训练力场”。

图注:在含910原子的体系上,Skala(蓝线)保持O(N³)斜率,而混合泛函(橙线)已显颓势
立即体验:
-
Azure云平台免费试用 -
GitHub开源代码 -
arXiv论文
当深夜的小王再次运行计算时,屏幕上跳动的数字或许正预示着——那个困扰化学家60年的”神圣泛函”,可能就藏在神经网络的权重矩阵里。